
When customers order oligonucleotides from Integrated DNA Technologies (IDT), they consistently receive high-quality oligos. Through improvements to traditional synthesis chemistries and advances in our proprietary synthesis platform, we can synthesize longer oligos with better sequence fidelity and a higher percentage of full-length product than competitors. Our technologies empower us to produce other synthesis products that can be used in many molecular biology applications. Here, we walk through our oligo manufacturing process and highlight the key factors that contribute to our industry-leading products.
Quality starts with proprietary synthesis platforms
The exceptional quality of IDT products starts with the equipment we use during synthesis. While many oligo manufacturers use the same general synthesis of protecting groups as described in Figure 1, nearly everything IDT uses to produce an oligo is made within our own facilities. We formulate all the key synthesis reagents in-house, and we design, manufacture, and calibrate our own DNA synthesizers and manufacturing software. This vertical integration enables us to internally monitor equipment performance, make quick alterations, and improve each step of the oligonucleotide synthesis process. This is an important advantage as it means IDT rarely relies on third party suppliers which simultaneously reduces the risk of supply chain delays.
IDT has engineered multiple synthesis platforms to accommodate the diversity of our customers' needs. For common, unmodified oligos, we use highly automated, high-throughput synthesizers ideal for rapid production and quick turnaround times. For nonstandard oligos, we use synthesizers optimized for enhanced flexibility. These synthesizers facilitate the addition of complex modifications, such as fluorophores, quenchers, linkers, spacers, and modified bases.
Our proprietary solid support yields a highly streamlined synthesis process
Though our synthesis platforms vary in speed and flexibility, they all use solid-phase synthesis to produce oligos. This means oligos will be tethered to a solid surface while they are being made [1]. At IDT, we engineer our solid supports in-house and can easily change their properties to accommodate oligo synthesis at different scales. This is a key reason why IDT is an industry-leading supplier of high-quality oligonucleotides.
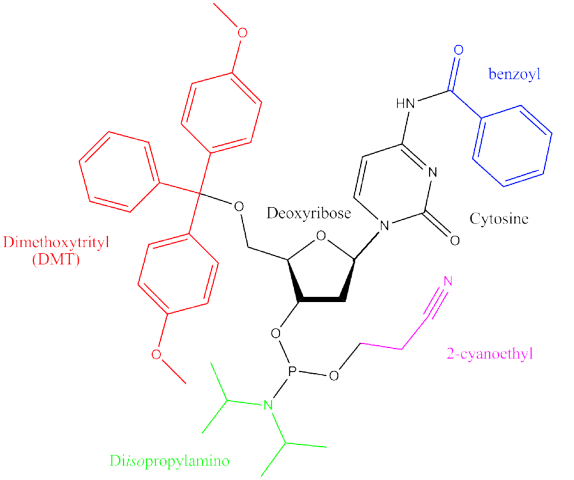
Before oligonucleotide synthesis, each solid support is fixed with a phosphoramidite monomer—a chemically-modified DNA nucleotide (Figure 1). The modified nucleotide is covalently attached to the solid support through its 3′ carbon. As oligonucleotide synthesis proceeds in the 3′→5′ direction, it becomes the 3′- most nucleotide of the oligo [1].
Note: Initial oligo subunits are not limited to nucleotides A, C, T, and G. IDT can also fix modifications to the solid support when they are needed at the 3′ end of an oligo.
What are phosphoramidites used for?
Phosphoramidite monomers, or phosphoramidites, are normal DNA nucleotides—adenine, cytosine, thymine, guanine (A, C, T, G)—that are chemically modified with protection groups. These protection groups prevent reactive amine, hydroxyl, and phosphate groups of the nucleotides from undergoing unwanted side reactions and force the formation of the desired product during synthesis. While the specific protection groups applied to each nucleotide vary among manufacturers, the use of a dimethoxytrityl (DMT) group for 5′ hydroxyl protection is ubiquitous in the industry (Figure 1). As we walk through the synthesis process in the following sections, we will use the term “nucleotides” in place of phosphoramidite monomers, but keep in mind this important distinction. Note too that phosphoramidites can be RNA nucleotides, including uracil (U).
A refined method for oligonucleotide synthesis
After a solid support containing the correct initial nucleotide (or modification) has been placed on the synthesis platform, IDT begins manufacturing the DNA or RNA oligo using a widely known method called phosphoramidite addition. Although many manufacturers employ this method, IDT uses chemical reagents that are formulated in-house to the standards of our unique manufacturing process. Several departments with IDT form a collaborative network of checks and balances, dedicated to ensuring efficient synthesis using precise chemistry and high-quality reagents. This commitment to quality provides our customers with consistent and reliable oligos.
The phosphoramidite approach: deblocking, coupling, capping, and oxidation
The synthesis process adds nucleotides one by one, using a repeated 4-step cycle of deblocking, coupling, capping, and oxidation for each A, C, T, or G addition. Figure 2, below, outlines this process.

Step 2: Coupling. After deblocking, the support-bound oligo is ready to couple with the next nucleotide in the oligo sequence. As a new nucleotide is introduced to the oligo, it reacts with a weak acid to form a “phosphoramidite intermediate”. This conversion allows nucleotide interaction with the unblocked hydroxyl group on the 5′ end of the receiving oligo, resulting in its covalent attachment through a phosphite triester bond.
Step 3: Capping. Despite many improvements to the phosphoramidite approach (see section below, A brief timeline of oligo synthesis), 100% coupling efficiency is not possible. Even with precise chemistry and pure reagents, some support-bound oligos will not couple with the added nucleotide, leaving a free 5′ hydroxyl group. If not blocked, this reactive group can couple to the nucleotide added in the next cycle, producing an oligo with a deletion in its sequence. To avoid this, a capping step is used to prevent any uncoupled molecules from further extension.
IDT caps the free 5′ hydroxyl groups by adding an acetylating reagent that renders them inert. Unable to participate in any subsequent reactions, capped oligos remain at their incomplete length for the duration of the synthesis process. The collection of capped oligos in the final sample—referred to as “truncated product”—can be removed by additional purification. While the presence of some truncated molecules in the final oligo product is inevitable for any oligo manufacturer, the goal is to minimize them. Our synthesis process consistently achieves extremely high coupling efficiencies (>99%), resulting in high quality oligos with low levels of truncated product (Figure 3).
Step 4: Oxidation. Once unreacted oligos have been capped, the phosphite-triester linkage between a growing oligo and its newly bound nucleotide must be stabilized. We accomplish this by adding a unique mixture of iodine and water into the synthesis platform. The mixtures oxidize the phosphite into phosphate, resulting in a stable phosphotriester bond between the oligo and its new nucleotide.
Repeated cycling, final processing, and desalting
Phosphoramidite addition repeats until the desired sequence is complete. Once the last nucleotide is attached, oligos undergo a process to remove the many protecting groups that are still attached. This includes a deblocking step to remove the DMT group attached to the final nucleotide, as well as other chemical reactions for the various nucleotide-specific protection groups. After these groups are removed, oligos are cleaved from the solid support, collected in a holding plate, and desalted to remove any additional contaminants. The final product is a functional single-stranded DNA molecule, ready for further purification (if needed), followed by IDT oligonucleotide quality control.
Oligo purification: PAGE and HPLC/UPLC
Long oligos, modified oligos, and oligos used in particularly demanding applications may require further purification to ensure that remaining truncated products do not compromise downstream reactions. IDT purifies such oligos using polyacrylamide gel electrophoresis (PAGE), high-performance liquid chromatography (HPLC), and/or ultra-performance liquid chromatography (UPLC).
PAGE. PAGE purification separates full-length product (FLP) from shorter species with great efficiency. It is most effective for unmodified oligos that only need truncated product removed. PAGE purification substantially reduces the amount (mass) of final oligo product, but most customers see the dramatic increase in purity as an acceptable trade.
RP- and IE- HPLC/UPLC. HPLC purification, a form of column chromatography, can be done one of two ways. Reverse-phase (RP) HPLC separates oligos based on their hydrophobicity, while ion-exchange (IE) HPLC separates oligonucleotides based on their charge. IDT uses HPLC for modified oligos that include additions such as linkers, spacers, non-standard bases, and hydrophobic modifications. The type of HPLC or UPLC used is dependent on the particular sample. Similar to PAGE, HPLC-purified oligos will undergo an unavoidable loss of mass, but this is offset by the gain in purity.
Regardless of length, IDT recommends that researchers consider additional purification (PAGE/HPLC/UPLC) for any oligo used in demanding applications such as site-directed mutagenesis, cloning, and gel-shift protein-binding assays. Our support team is available to provide purification recommendations. Contact us for assistance.
More information can be found on our DNA oligos and RNA oligos webpages.
IDT quality control: ESI-MS, CE, and UPLC
Oligo purification does not provide information about oligo quality. We understand how important it is to produce a product that delivers consistent results to our customers, which is why we are committed to ensuring that the oligos we produce are the correct sequence and include a high proportion of full-length product.
ESI-MS. For sequence verification quality control, IDT uses electrospray ionization mass spectrometry (ESI-MS). The technique measures the final mass of an oligo, which is then compared to the expected mass based on the specific oligonucleotide sequence. Thus, if an oligo is missing a nucleotide, contains an extra nucleotide, or contains an incorrect nucleotide, it will be evident here. While many oligo manufacturers use matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry to measure oligonucleotide mass, our extensive experience has shown that this method becomes less accurate for oligos >40 nt and is ineffective for long oligos >75 nt. ESI-MS, however, can accurately measure oligos up to 200 nt. All IDT oligos that do not contain mixed bases undergo ESI-MS for sequence verification, and we provide this quality control data to our customers with every order they place. Oligos with mixed bases represent a mixed population of oligo masses and cannot be accurately measured by ESI-MS.
CE. Capillary electrophoresis (CE) is a quality control measure we use to determine the percentage of full-length product relative to truncated product. The method allows us to monitor the performance of our synthesizers and purification methods and is also used to provide customers with purity guarantees. IDT provides complementary purity guarantees and CE analysis for purified, unmodified oligos <60 nt that have low secondary structure. For all other oligos, customers can receive CE analysis for their order by purchasing a purity guarantee separately.
Analytical HPLC. Certain oligos possess secondary structures that can lead to anomalous CE results. For these oligos, we use analytical HPLC to assess their purity—this can be done with both RP and/or IE UPLC, depending on the oligo’s properties. Additionally, IDT will use analytical HPLC for quality control on other oligos if specifically requested by the researcher.
IDT coupling efficiencies show industry-leading consistency
Chemical and physical restraints reduce coupling efficiency to <100% during each cycle of synthesis (see The phosphoramidite approach section, Step 3: Capping). As a result, the small percentage of truncated product (<1%), generated with every nucleotide addition, accumulates. Thus, as sequence length increases, so does the total percent of truncated product. A 20mer oligo with 99.4% coupling efficiency will have an estimated 89.2% FLP, while a 50mer with the same coupling efficiency will have an estimated 74.5% FLP. The relationship between oligo length, coupling efficiency, and FLP percentage is shown in Figure 3.

Figure 1. IDT proprietary platforms have a better coupling efficiency than other suppliers, which provides more full-length oligonucleotides in your order. Small increases in coupling efficiency (≤1%) result in measurable increases in full-length product yield. The curves represent the theoretical yield for different lengths of oligos based on a coupling efficiency of 99.4% (IDT oligos, n = 126) and 99.1% (other supplier, n = 134 from three different suppliers) using the formula, percent full length product = (eff )(n-1)*100 where eff = coupling efficiency (for example, 99.4% = 0.994) and (n–1) is the number of coupling reactions needed to make an oligo of length n.
For long oligos, IDT offers an enriched synthesis cycling and proprietary solid support that has an even higher coupling efficiency of 99.6% (Figure 4). IDT Ultramer™ DNA Oligos are suggested for any customer requiring oligos between 45 and 200 bases in length. IDT also uses this proprietary synthesis technology to manufacture oPools™ Oligo Pools. These are combinations of custom, single-stranded DNA sequences for creating CRISPR libraries, primer pools for multiplex PCR, data storage, or gene construction. Because of the highest coupling efficiency, oPools can be ordered for oligos up to 350 nt in length, the longest pooled oligos available on the market.

Figure 4. IDT proprietary platforms have a better coupling efficiency than other suppliers, which provides more full-length oligonucleotides in your order. Small increases in coupling efficiency (≤1%) result in measurable increases in full-length product yield. The curves represent the theoretical yield for different lengths of oligos based on a coupling efficiency of 99.6% (IDT oligos, n = 34,627) and 99.4% (other suppliers, n = 77 from three different suppliers) using the formula, percent full-length product = (eff)(n–1)*100 where eff = coupling efficiency (for example, 99.4% = 0.994) and (n–1) is the number of coupling reactions needed to make an oligo of length n.

IDT: A global leader in nucleic acid synthesis
Due to our proprietary manufacturing processes and dedication to quality control, IDT is a major global supplier of custom nucleic acids. All based on the oligo synthesis technologies described here, IDT products support a wide variety of applications, including next generation sequencing (NGS), DNA amplification, genotyping and SNP detection, genome editing, expression profiling, gene quantification, synthetic biology, and functional genomics.
Today, IDT manufactures more than 90,000 oligos each day and has served more than 130,000 researchers around the globe. IDT manufacturing locations include facilities in Coralville, Iowa; Research Triangle Park, North Carolina; San Diego, California; Leuven, Belgium; and Singapore.
